-
Research
-
Publications
-
All publications
-
Benner, SA
-
Biondi, E
-
Bradley, K
-
Chen, C
-
Hoshika, S
-
Karalkar, N
-
Kim, HJ
-
Kim, MJ
-
Laos, R
-
Leal, NA
-
Li, Y
-
Richards, N
-
Shaw, RW
-
Spacek, J
-
Yang, ZY
-
People
-
Benner, Steven
-
Biondi, Elisa
-
Bradley, Kevin
-
Chen, Cen
-
Darling, April
-
Hoshika, Shuichi
-
Karalkar, Nilesh
-
Kim, Hyo-Joong
-
Kim, Myong-Jung
-
Laos, Roberto
-
Leal, Nicole
-
Li, Yubing
-
Richards, Nigel
-
Shaw, Ryan
-
Spacek, Jan
-
Yang, Zunyi
-
News and Events
-
Press Coverage
-
Our Foundation
|
Medical Genomics

Small molecule therapeutics
FfAME technologies are also the foundation for the development of new classes of therapeutics. In parallel with research in the laboratory of Nobel Laureate Jean Marie Lehn, our group invented Dynamic Combinatorial Chemistry as a drug discovery technology. This technology supported the development of Alantos, a pharmaceutical company that was later sold to Amgen. As an example of small molecule evolution, dynamic combinatorial chemistry was also used in products developed by FfAME scientists to assemble primer combinations for use in diagnostics and genomic analysis.
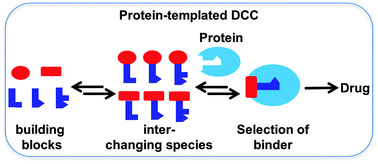
Dynamic combinatorial chemistry allows a drug target to select its own favorite drug.
Large molecule therapeutics
An emerging concept in cancer therapy uses antibodies conjugated to toxic drugs via digestible linkers, keeping the drugs inactive until the antibody-drug-conjugate (ADC) is taken up by a targeted cancer cell. While ADCs have advantages in broadening the therapeutic window of a drug that, if free in solution cause dangerous side effects, they also have disadvantages. These include their long development time, low drug loading, large dosing times, and very high cost. The ability to evolve AEGIS has allowed us to evolve molecules that bind selectively to cancer cells. These "AEGISbodies" can replace antibodies in the ADC concept, saving 99% of the cost ($20,000 vs. $200 per dose) while giving higher drug loads, better control over dosing, shorter dosage times, and shorter development times. With further development of the technology funded by the NIH, we are now seeking collaborators to move drug delivery systems that exploit AEGISbodies into preclinical studies.
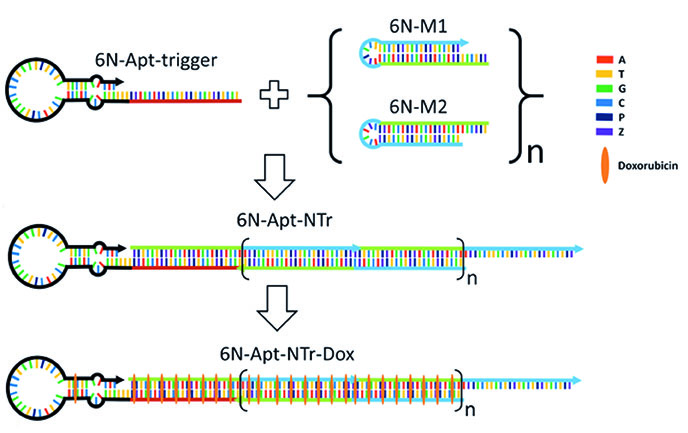
A drug delivery nanostructure built from 6 different nucleotides (A, T, G, C, and AEGIS P and Z) delivers toxic docorubicin selectively to liver cancer cells.
Today, with Firebird, TrakItNow, Genepath, and the Florida Medical Entomology laboratory, a team at FfAME is applying our innovations in environmental surveillance and human diagnostics in low research environments, such as rural India. This work is now supported by the CDC and the binational USISTEF. The National Institute of Allergies and Infectious Disease is presently supporting a FfAME program to develop the basic science behind molecular diagnostics.
In the recent COVID pandemic, the experience of the group with SARS in 2003 allowed us in January 2020 to predict that COVID was going to be "the big one", the long feared global pandemic. The group's chemical innovations were immediately used to create a COVID diagnostics test. When the COVID test designed by the CDC failed, our test was moved towards regulatory approval. The Indian government approved a PCR test with Genepath that incorporated FfAME's technology six weeks after it began review, with US FDA approval following in August. Later, with Sparsek, a Czech company, European regulators approved a 15-minute sample-to-results test incorporating FfAME's reagents, this one using proprietary isothermal displaceable probe loop amplification technology.
FfAME technology was incorporated in tests to quickly diagnosis SARs-CoV2.
|
|
|


